来源:MedTrend 医趋势 发表日期:2018年6月5日
FDA局长Scott Gottlieb从2017年5月走马上任后,一直在加速药物审批程序,比如颁布「突破性疗法」认证程序将审查时间减少到3个月,缩短将近1/3的时间,2017年新药审批数创21年来新高,有46种新药获批,开启“以患者为中心”的药物经济价值预测时代。
近日,在芝加哥举行的美国临床肿瘤(ASCO)年会上,Scott Gottlieb透露FDA或将颁布重大创新性肿瘤新药审批新政策——“实时审评制”。肿瘤新药上市审批或将提前到临床阶段,完成临床试验后可直接上市,无需排队审批。
颠覆式打破抗癌新药审评监管中的障碍,对于药企来说无疑是重大利好消息!
▲FDA局长Dr. Scott Gottlieb,图片来源Cameron Costa | CNBC
什么是“实时审评制”?
“实时审评制”的具体流程是根据药企提供的肿瘤新药做一份共享申请表格,允许FDA审查员将评论添加到这些背景文件中,确保信息及时沟通和分享。
在提交申请表和被批准之前,FDA需要预先和实时审议临床试验数据:
▪ 一是检查药企提供的数据完整性和是否齐备;
▪ 二是打破以往中规中矩的审评流程,帮助药企解决试验药品的质量问题。
也就是说,原先的先进行临床试验再进行新药申请审批,转为边进行临床试验边进行新药审批。
肿瘤新药审批提速至少半年!惠及所有癌症新药
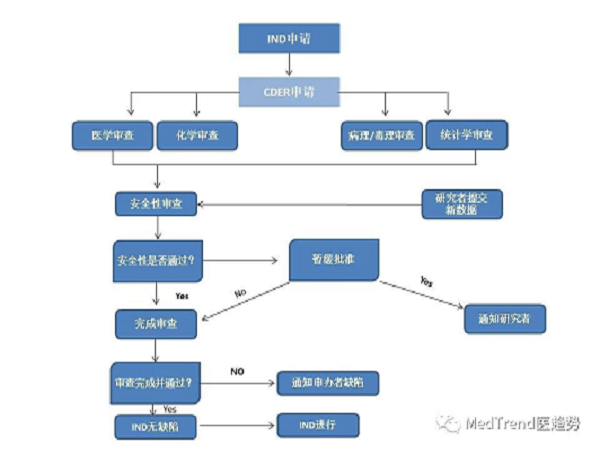
以往新药研发到上市流程
按照原来新药研发到上市的流程,可分为:
临床前研究:研究开发(2-3年)、临床前试验(2-4年)
临床试验审批(IND):(1个月以上)
临床试验(一般3-7年):
人体试验共分三期:
Ⅰ期临床 20-100例,正常人,主要进行安全性评价。
Ⅱ期临床 100-300例,病人,主要进行有效性评价 。
Ⅲ期临床 300-5000例,病人,扩大样本量,进一步评价。
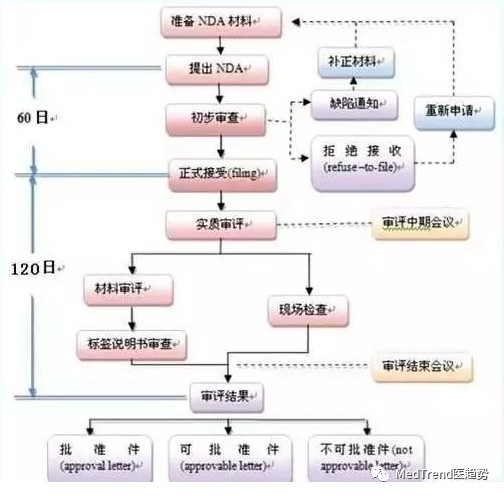
药上市审批(NDA):半年左右
上市后研究:临床监测期——Ⅳ期临床,受试者大于2000例;
上市后再审批(上市后4-10年):重新审核 NDA 中的有效性和安全性。
传统的药物审评,80%的审查都集中在临床部分,20%会专注产品自身的问题。所以,一款肿瘤新药从研发到上市,期间起码需要花费十几年的时间。许多新药研发在临床1期、临床2期就已经失败,能够进入临床3期阶段的寥寥无几。
而原来药企在做完临床3期试验后,才能提交报告去排队等候FDA审批批复,根据2017年的标准审评时间为17个月左右,而回答问话、补充相应材料都花费大量时间。
“实时审评制”缩减新药上市时间
如果实行“实时审评制”后,药企在进行临床试验阶段,FDA就开始参与试验数据审评,随时在企业做实验的过程中告知需要改进之处,或者补充材料及补做试验,如果数据不合格,或在临床1期、2期阶段就将药品淘汰,避免药企后续繁复的试验和申请工作。
而完全临床3期阶段时,药企或将跳过审批直接上市,对药企来说,具有颠覆性意义。大大缩短了新药上市时间,预计至少提速半年时间,也让肿瘤患者能更快获得更优质的药物治疗。
目前,“实时审评制”已在上市肿瘤药扩大适应症申请中试点应用。如果试点成功,FDA或将会把此审评方法和流程扩展到所有癌症新药上市申请程序中去。
此外,除了肿瘤药,FDA也有意简化基因治疗和细胞治疗的审查流程。
Scott Gottlieb表示,FDA正在进行细胞和基因治疗的新型临床试验设计的应用科学研究,并且通过监管制定药物加快开发计划。包括使用“突破性疗法”认定,以及再生医学先进疗法认定(RMAT)。
FDA多渠道加速审批提速,两款CAR-T疗法从中受益
早在2017年底,FDA就宣布将对特定癌症药物审批提速。
▪ 一是对已经获批的一个适应症癌症疗法,通过递交基于“更有针对性的数据集(如单臂研究)”的补充申请,获得第二个适应症的批准。
▪ 二是根据中间临床终点(intermediate clinical endpoints)加速批准新药上市。
两个突破性CAR-T疗法:诺华的Kymriah和Kite的Yescarta都分别获得了FDA优先审评资格和快速通道资格,都成为FDA新药审批提速的受益者。
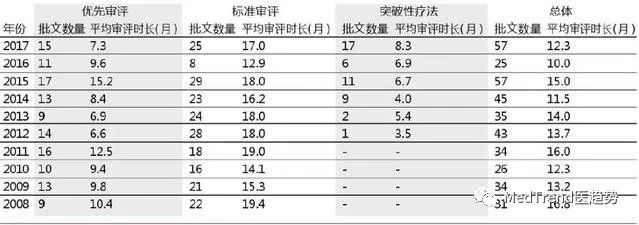
▲不同审评方式时长对比
根据上表显示,2017年标准审评时间为17个月,而进入优先审评的药物为7.3个月,突破性疗法的审评时间为8.3个月。相比标准审评时间减少10个月左右。Kymriah和Yescarta这些优先审评路径下的药物获批时限约为7.3个月,达到2013年以后的最快速度。
如果实施“实时审评制”,CAR-T疗法连这7.3个月的审评时间或将免去!未来CAR-T疗法竞争也将越来越激烈。
▲FDA批准的3种基因疗法(信息来源:FDA官网)
在基因疗法方面,去年就有三种基因疗法获得批准: 两种治疗血癌基于细胞的基因疗法,一种治疗遗传性视网膜萎缩症的直接型基因疗法,将基因疗法从理想变为现实。但这三类基因疗法产品只是冰山一角。
▲2017年11月发布的再生医学框架中提到将采取5种途径加快再生医学产品审批(图片来源:FDA官网)
正如前文所说的,FDA对特定药物自2016年12月开始,启动再生先进疗法认定(RMAT)。截至2018年4月底,共有62份RMAT认定材料提交,FDA颁发了19个认定。在这19种产品中,有14种也获得了罕见药认定。表明该计划促进了罕见疾药物的开发。
麻省理工学院发表的一篇论文基于2017年的930种在研产品线预测。到2022年底,将有约40种基因疗法产品获得FDA批准。此外,这些批准的基因疗法中45%都将用于治疗癌症。
一直以来,FDA一面顶着快速批准药物的压力,一面希望获取令人信服的临床数据,努力在二者之间达成平衡。
如果FDA“实时审评制”实施,或许有助于实现这种平衡。让药物研发在前中期就能基于合理预测的审评进行实时数据调整,帮助新产品批准后能对其安全问题进行更多实时监控。
未来,肿瘤新药研发上市速度或将创新高,更多肿瘤新药和疗法的竞争也将越来越激烈。更多新药的上市,对于也给了患者一个摆脱病魔的更多机会。