来源 来源:药时空
摘要
抗体药物作为作为一种高剂量使用的生物制剂对产品纯度以及杂质残留量要求非常严格另上游表达量越来越高产品要求也越来越高随之而来的是下游纯化压力。如何快速获得抗体的纯化产品以及优化其生产工艺成为药物研发公司关注的要点。本篇文章介绍了抗体药物发展史以及下游纯化工艺的基本流程主要包括深层过滤低pH灭活中间品深层过滤离子层析除病毒过滤和超滤渗滤。
一 单克隆抗体简介
抗体是一类免疫球蛋白ImmunoglobulinIg当母体受内源或外源物质刺激时B淋巴细胞特异性识别抗原决定簇从而促使一系列分化和分裂行为B细胞的终末分化细胞即浆细胞合成了抗体。每一个B淋巴细胞只能分泌一种识别、结合抗原决定簇的抗体。单克隆抗体(Monoclonal antibodiesMAbs)是由B淋巴细胞和骨髓瘤细胞杂交形成的杂交瘤细胞产生的具有高度均一性和特异性。
MAbs由两条重链与两条轻链构成[1]其中重链分子量约为50~75KD含约450~550个氨基酸由一个可变区域V和三或四个恒定区域C组成具有介导重要的免疫学功能高等脊椎动物含有五种类型的重链γμαδε分别决定球蛋白的五种类型IgGIgMIgAIgDIgE[2]而轻链分子量为25KD含大约214个氨基酸由一个可变区域V和一个恒定区域C组成具有参与结合抗原并稳定免疫球蛋白结构的功能。一个二硫键将其中的一个重链和轻链连接起来这些二硫键处在一个被称为铰链区(hinge)的柔性部位上这个区域由12个氨基酸组成且易于被蛋白酶或化学物质所切割。IgG可被木瓜蛋白酶水解为两个相同以及一个不同片段两个相同片段在溶液或凝胶中不能形成片段且以单价形式与抗原结合因此被称为Fab 片段Fragment antigen binding抗原结合片段另一个不同片段不能与抗原结合且比Fab更容易产生结晶其结构有更高的均一性被称为Fc片段Fragment crystallisable可结晶片段[3]。抗体的重链以及轻链的可变区VH+VL称为Fv片段Fragment variable不稳定片段是一个能够结合抗原的不稳定片段。可变区可被细分为高度可变区或者CDRscomplementarity-determining regions互补决定区该区域直接和抗原或框架区域用作CDR与抗原接触的支架结合[4]。如下图1-1
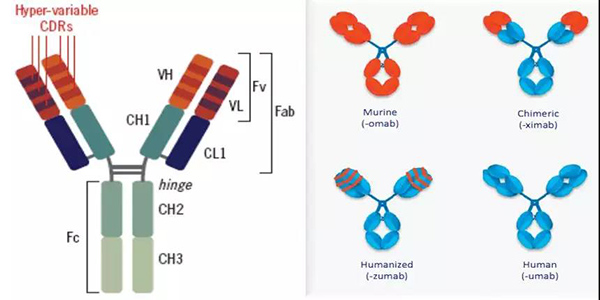
图1-1 单克隆抗体示意图 图1-2 单克隆抗体类型
Kohler和Milstein两位科学家于1975年发明了单克隆抗体技术[5]也因此获得了1984年的诺贝尔医学奖。随着基因工程技术的迅速发展治疗性MAbs从早期100%的鼠源MAbs到人-鼠嵌合抗体、人源化抗体再到全人源性抗体如图1-2并衍生出双特异性抗体[6]纳米抗体等多种新型抗体药物逐渐解决了异源性抗体的免疫原性问题。以下是针对鼠源抗体到新型抗体药物发展的相关介绍。
1.鼠源抗体(Murine antibody)使用异种抗原免疫小鼠然后通过纯化小鼠腹水得到针对该抗原的多克隆抗体再经过骨髓瘤细胞融合得到可持续表达的单克隆抗体。1986年FDA批准的全球首个单抗Orthoclone-OKT用于抗移植排斥的治疗[7]。但是鼠源抗体可诱导产生HAMAHuman Anti Mouse Antibody人抗鼠抗体使鼠源抗体加速清除增加病人后续应用的临床风险。
2.人-鼠嵌合抗体Chimeric antibody恒定区来自人IgG可变区来自鼠MAbs是一个人-鼠杂合的抗体。该抗体不仅能大幅度降低异源抗体的免疫源性其人源Fc片段还能有效介导生物学效应功能如CDCComplement dependent cytotoxicity补体依赖性细胞毒作用ADCCAntibody-dependent cell-mediate dcytotoxicity抗体依赖细胞介导的细胞毒作用[6]等。
3.人源化抗体(Humanized antibody)小鼠CDRs与人的抗体框架嫁接经亲和力重塑保留亲本鼠单克隆抗体的特异性和亲和力几乎完全去除免疫原性和毒副作用。
4.全人抗体(Human antibody)通过转基因技术 将人类编码抗体的基因全部转移至基因工程改造过的抗体基因缺失的动物中从而使动物表达人类抗体达到抗体全人源化的目的。
5.双特异性抗体Bispecific antibody一种人造蛋白由于重链与轻链来源于不同的抗体所以可以与两种特异的抗原结合。双特异性抗体的优势表现在增强了肿瘤细胞的杀伤力可以同时阻断两个不同信号路径通过两个不同细胞表面抗原相互作用增加特异性结合[8]。
二. 国内外单克隆抗体研究进展及发展趋势
MAbs药物因其靶向性强、特异性高和毒副作用低等优势迅速在肿瘤和自身免疫病等领域得到广泛应用市场规模成长迅速。据EvaluatePharma统计全球在研抗体药物占所有在研生物药的70%。至此FDA共有38个治疗用抗体药物其中包括Fc和Fab融合蛋白被批准上市其中有2个抗体由于临床在批准上市后退市。2011年Top50生物药物中有14个为抗体药物前6位全部是抗体药物年销售额均超过50亿美元。其中第一位是abbott公司的Humira2011年销售额达到79亿美元。FDA批准的MAbs类药物中40%用于治疗癌症33%用于治疗自身免疫10%用于器官移植治疗5%用于治疗感染12%应用于其他疾病。虽然生物技术制药市场还比较小例如在2011年生物技术制药只占全球药物生产市场的15%但是抗体药物的复合增长率却很高为40%。同时各大原研单抗的专利陆续到期EMAEuropean Medicines Agency欧洲药品局和FDA生物仿制药申报法规的陆续出台为单抗类生物仿制药的井喷式发展提供了绝佳窗口。
中国的MAbs产业起步较晚研发工艺能力与发达国家存在巨大差距至2016年本土研发生产并上市的单抗及Fc融合蛋白类产品仅10个。国内MAbs药物行业面临的主要问题有
1.动物细胞大规模培养表达量远低于国外水平。国内细胞大规模的表达量一般在
0.5-1g/L并且工业化生产规模小最大的为3000L。而国外上市的药物的表达量在2g/L左右临床的表达量在3-5g/L之间
2.MAbs纯化工艺水平较低。FDA在2005年已经要求使用QbDQuality by Design,质量源于设计的研究方式研究生产工艺。现在国内仅有少数研发企业使用QbD的模式而且多数企业使用传统的研究方法。如何降低工艺成本提高收率提高工艺稳定性在抗体药物研发中是非常重要的。
差距意味着潜力和机遇国内肿瘤发病率持续上升构成的庞大市场需求极大刺激了本土企业开发 “新药创制重大专项”在公布的2016年计划资助100多个项目中单抗项目数占16%。据不完全统计截止2016年11月国内共有超过90家企业涉足单抗药物研发相关研发项目超过280个。不久后的中国单抗市场注定会呈现爆发式增长局面预计2020年将有6-10个新的本土单抗被推向市场。
近年来是MAbs发展的黄金时期但是还需要面对另一个客观事实目前国际上针对热门靶点的单抗项目众多比如针对TNF的项目超过40个VEGF/VEGFR单抗项目超过35个HER2超过25个。因此上市速度将会是决定产品市场地位的一个重要因素而以高产能效率、高灵活性、低生产成本为主要特点的一次性技术产品也必将在未来的生物工艺解决方案市场大放异彩。近几年上市的MAbs体现了下一代MAbs药物研发趋势
1.减小MAbs分子量从而更有利于药代动力学以及药效学参数改善制剂的属性更利于生产。例如单链MAbs以及MAbs的结构域。
2.通过已知的药物的分子模型创造新的药物。两个或两个以上的生物功能分子形成融合蛋白MAbs片段形成多价或多特异性。例如双特体MAbs。
3.提高新一代的MAbs的生产能力以及其在生产过程中的稳定性和产品的稳定性。
4.提高药物的关键参数如MAbs的半衰期和体内分布免疫的安全性。
5.提高药理作用即目标的特异性和结合特性包括亲和力和结合动力学。
三.单抗纯化工艺与优化
1.三步纯化策略
在每一个生物技术工艺中生物反应阶段后要考虑进行两个子工艺即回收和纯化这两步操作也被称为下游工艺。回收步骤包括培养基分离、细胞破碎、残片去除等初步单元操作。残留的产品实际上是包含目标蛋白且具有不同物理化学性质的蛋白复合物 [9]。可见从如此混杂的蛋白中提取一种目的蛋白是一件非常艰巨的工作。故蛋白纯化工艺应建立在良好的蛋白理化性质基础之上合理并充分利用处理技术方法和参数尽量优化工艺步骤设计一系列高效低耗和易于实行的蛋白质纯化分离方案[10]。
在生物制药产业三步纯化策略用于帮助治疗性蛋白纯化工艺的开发并且在实验室开发纯化方法时同样有效。注意三步纯化策略并不是指所有的纯化策略都应该包括这三部纯化步骤。
1捕获粗纯
该步骤目的是分离、浓缩、稳定目标蛋白。产品被浓缩转移到一个可以维持其效能、活性的环境。最好可以去除其他关键杂质。
2.中间产品纯化
该步目的是去除大部分性质相似的杂质例如其他蛋白质和核酸、内毒素和病毒。
3.精制
这一过程目的是去除一些微量的和分子量相近的杂质和多聚体。
2.单抗纯化操作流程
下游工艺先通过澄清发酵液捕获MAbs再通过一系列层析单元操作组合纯化MAbs。该过程中还包括专门用于病毒灭活和去除的两个正交步骤。然后通过渗滤和超滤步骤将抗体制成适于药物产品制造的混合物和浓度。制成的产品经0.2μm除菌过滤器过滤装入适当的容器中并冷冻保存。MAbs下游纯化过程包括8个步骤如图2-1

图2-1 蛋白纯化流程
1.深层过滤Depth FiltrationDF
也称澄清过滤属于流体通过多孔介质的渗流[11]用于系统清除残留的固体和较小的细胞碎片。深层过滤器通过两种主要机制确保颗粒和溶质的正常流动分离筛分和吸附。基本过程分为两个阶段第一阶段是输送阶段悬浮颗粒在多种力的作用下向滤料表面迁移[12]第二阶段是附着阶段颗粒在各种物理化学力、流体剪切力、碰撞力等的作用下附着在滤料表面并被截留、捕获。其高容量是由于污染物被捕获并保留在介质的整个深度中而不是仅被保留在过滤器的表面上[13]。高表面积和带电相互作用的可能性意味着深层滤器除了具有捕获较大颗粒的能力之外还具有吸附性能[14]。
2.亲和层析Affinity ChromatographyAC
亲和填料含有一种名为Protein A的配基。Protein A一种从金黄色葡萄球菌细胞壁中获得的蛋白质具有与免疫球蛋白选择性相互作用的能力IgG通过氢键和两个盐桥的疏水相互作用使其Fc区与蛋白A结合[15]。
AC是单抗纯化第一步可使蛋白纯度达到95%以上[16]。该步骤目的是从澄清的收获液中捕获单克隆抗体同时去除工艺相关杂质HCPHost Cell Protein宿主细胞蛋白DNA和小分子杂质。
3.低pH灭活
也称病毒灭活灭活可能存在于哺乳动物细胞培养物中的治疗性蛋白质产品中的脂包膜病毒。脂包膜病毒的主要功能是帮助病毒进入宿主细胞。在pH3.9及以下可以有效灭活脂包膜病毒pH越低灭活病毒效果越好。
4.阳离子层析Cation Exchange ChromatographyCEX
离子交换色谱分离生物分子的基础是待分离物质在特定条件下与离子交换剂带相反电荷因而能够与之竞争结合[17]。该步骤的目的既将多聚物蛋白质聚集体对治疗性蛋白质产物的免疫应答构成风险[18]降低至药物的可接受水平又将HCP降低到可接受的水平以便后续的阴离子层析处理。
5.阴离子层析Anion Exchange ChromatographyAEX
该步骤目的是去除HCP、DNA、Protein A和内毒素达到药物的水平物质验收标准。同时也起到病毒清除的作用。
6.纳滤NanofitrationNF
为了保证产品安全潜在的病毒污染物通过使用传统上被认为可靠的用于病毒清除的纳米过滤器去除[19]。NF去除可能存在于哺乳动物细胞培养物产物中的细小病毒如小鼠微小病毒minute virus of miceMVM和较大病毒如小鼠白血病病毒murine leukemia virusMuLV。纳米膜是一种功能性的半透膜[20]NF只与目的蛋白和病毒分子的大小有关依据蛋白分子量和膜截留量大小选择合适孔径的膜以压力差为推动力从而达到显著去除病毒的目的[21]。
7.超滤/渗滤UltrafiltrationUF/ DiafiltrationDF
UF基于膜孔径或分子量截留分离溶液中的分子如图2-2。DF常用于在恒定体积下改变回流溶液的化学性质如图2-3。 不需要的颗粒通过膜同时通过添加置换缓冲液将原料液的组成改变为更理想的状态。在生物技术中渗滤通常用于将产物交换到最终制剂缓冲液中[22]。UF和DF常使用切向流过滤[23]其中料液平行流过膜表面而过滤的同时也对滤膜进行了冲刷使膜表面不会形成凝胶层从而使料液中的颗粒不会很快堵塞滤膜。该步骤目的是确保单克隆抗体制剂和浓度达到药物规格。
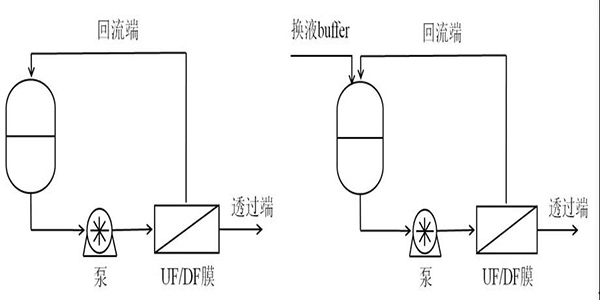
图2- 2 超滤装置 图2- 3 渗滤装置
3.优化方法
1.实验设计Design of ExperimentDoE
DoE顾名思义就是设计实验的过程用于收集易于用统计方法分析的数据从而得出有效客观的结论[24]。实验设计在科学和工程学领域是改进产品实现过程非常重要的手段也是制造过程设计与开发和过程管理中的重要元素。我们可把实验看做科学研究的一部分或将其视作过程运行方式和探究系统的一种途径。一般来说先针对某一过程提供一些猜测接着进行实验操作然后产生这一实验的相关数据再利用来自这一实验的信息设立另一个新的猜测这一新的猜测又导出另一个新的实验。
该技术允许我们通过少量的实验获得足够多的数据只需要在实验过程中系统的改变实验因素。基于所获得数据可以建立一个用于研究工艺的数学模型以此获得实验因素对结果的影响并找到最佳的工艺条件。可借助现代软件创建DoE、获取数学模型、使产生的信息可视化。DoE常用的方法是确定一个中心点然后围绕改点进行代表性的实验。DoE可以极大地提高筛选合适的实验条件的效率。
2.高通量筛选High Through ScreeningHTS
为了提供一个简单的用来优化纯化MAbs的工具人们在建立DoE与96孔板实验结合的基础上开发了HTS方法[25]。96过滤孔板可以根据实验者的实验设计需求自由填充不同微量的填料孔板中层析填料和目标蛋白之间的基本相互作用和层析柱中是一样的。HTS应用于早期层析筛选实验既节省时间节约成本又加速工艺开发和优化进程[26]。工艺开发过程中的HTS技术可以允许大型实验空间的表征、支持已经建立的良好的设计空间的确认从而控制被监测的关键工艺参数、减少临床时间。
总结
抗体在下游纯化工艺中主要经过捕获富集中度纯化去除大部分杂质精细纯化去除一些微量的和分子量相近的杂质根据目的不同从而获得较高纯度的产品。
参考文献
[1] 于传飞, 王文波, 李萌, 等. 人源化抗VEGF单克隆抗体制品的大小异质性分析[J]. 中华微生物学和免疫学杂志, 2014, 34(009): 718-722.
[2] Wilson K, Walker J. Principles and techniques of practical biochemistry[M]. Cambridge University Press, 2000.
[3] Spadiut O, Capone S, Krainer F, et al. Microbials for the production of monoclonal antibodies and antibody fragments[J]. Trends in biotechnology, 2014, 32(1): 54-60.
[4] Buss N A P S, Henderson S J, McFarlane M, et al. Monoclonal antibody therapeutics: history and future[J]. Current opinion in pharmacology, 2012, 12(5): 615-622.
[5] 潘萌, 孔蕴, 陈畅, 等. 单克隆抗体药物的研究进展[J]. 中国生化药物杂志. 2008, 29(1): 62-64
[6] Ribatti D. From the discovery of monoclonal antibodies to their therapeutic application: An historical reappraisal[J]. Immunology letters, 2014, 161(1): 96-99.
[7] Liu J K H. The history of monoclonal antibody development–Progress, remaining challenges and future innovations[J]. Annals of medicine and surgery, 2014, 3(4): 113-116.
[8] Fan G, Wang Z, Hao M, et al. Bispecific antibodies and their applications[J]. Journal of hematology &>[9] Vásquez‐Alvarez E, Lienqueo M E, Pinto J M. Optimal synthesis of protein purification processes[J]. Biotechnology progress, 2001, 17(4): 685-696.
[10] 张建社, 禇武英, 陈韬. 蛋白质分离与纯化技术[M]. 军事科学出版社, 2009.
[11] 康勇, 罗茜. 液体过滤与过滤介质[M]. 化学工业出版社, 2008.
[12] 李孟, 刘新明. 国内外深层过滤理论的最新研究进展[J]. 中国水运, 2006, 6(10): 45-46.
[13] Merdy S L. Selection of Clarification Methods for Improved Downstream Performance and Economics[J]. Bioprocessing Journal, 2015. 14(1): 1538-8786.
[14] Yigzaw Y, Piper R, Tran M, et al. Exploitation of the adsorptive properties of depth filters for host cell protein removal during monoclonal antibody purification[J]. Biotechnology progress, 2006, 22(1): 288-296.
[15] Hahn R, Schlegel R, Jungbauer A. Comparison of protein A affinity sorbents[J]. Journal of Chromatography B, 2003, 790(1): 35-51.
[16] 陈泉, 卓燕玲, 许爱娜, 等. 蛋白A亲和层析法纯化单克隆抗体工艺的优化[J]. 生物工程学报, 2016(1), 6: 010.
[17] 周楠迪, 田亚平. 《生物分离原理与技术》卓越课程建设的探索[J]. 教育教学论坛, 2014 (13): 198-199.
[18] Miesegaes G R, Lute S, Strauss D M, et al. Monoclonal antibody capture and viral clearance by cation exchange chromatography[J]. Biotechnology and bioengineering, 2012, 109(8): 2048-2058.
[19] Vázquez‐Rey M, Lang D A. Aggregates in monoclonal antibody manufacturing processes[J]. Biotechnology and bioengineering, 2011, 108(7): 1494-1508.
[20] 彭跃莲. 膜技术前沿及工程应用[M]. 中国纺织出版社, 2009.
[21] 张翠萍, 周安, 杨虎虎, 等. 人抗凝血酶Ⅲ纳米膜过滤工艺优化[J]. 中国输血杂志, 2015, 28(12): 1481-1484.
[22] Houp R C. Ultrafiltration and diafiltration[J]. Journal of Validation Technology, 2009, 15(4): 40.
[23] Miao F, Velayudhan A, DiBella E, et al. Theoretical analysis of excipient concentrations during the final ultrafiltration/diafiltration step of therapeutic antibody[J]. Biotechnology progress, 2009, 25(4): 964-972.
[24] Douglash C.Montgomery. 实验设计与分析: 第6版[M]. 人民邮电出版社, 2009.
[25] Healthcare G E. High-throughput screening and optimization of a multimodal polishing step in a monoclonal antibody purification process[R]. Application note 28-9509-60 AB, 2009.
[26] 孙文改, 苗景赟. 抗体生产纯化技术[J]. 中国生物工程杂志, 2008, 28(10): 141-152.
如涉及知识产权请与我司联系
|


