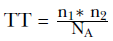如何确定新药人体内首次给药(first in human,FIH)剂量是新药研究过程中的重要环节之一,在临床实验的过程中,FIH剂量代表着临床实验的起点,因此必须绝对安全。在FDA的指导方案中,FIH可以通过在相关物种上进行的多种药理、毒理研究得出的,“未观察到不良反应水平”(No Observable Adverse Effect Level,NOAEL)确定,此概念一般认为是临床前药理毒理实验中,在相关物种上确定的最大安全剂量。通过物种敏感度、特异性及人体适用性等相关因素可以确定NOAEL的人体等效剂量(human equivalent dose,HED),再加上特定的安全系数(safety factor)计算得出人体最大安全起始剂量(human maximum recommended safe starting dose,MRSD)。然而,能够高度激活免疫系统的生物药物,即使在低剂量下也可能会引起诸如细胞因子释放综合征、神经系统毒性等强烈的副作用。
在TGN1412,CD28超级激动剂临床实验Ⅰ期“惨案”过后,为了提高临床实验的安全性,EMA和FDA在2007年发布了针对生物药FIH确立的具体指导方针,其中强调了包括双特异性抗体在内的一系列药物的FIH计算,应该基于“最小预期生物效应级别”(minimum anticipated biological effect level,MABEL),此概念认为是在人体中可以被认为有生物学效果的最小剂量。
近日,来自杜克大学的学者在《Journal for Immunotherapy of Cancer》上发表的文章,详细分享了通过MABEL方法计算他们设计的一种用于治疗胶质母细胞瘤的EGFRvⅢ-CD3双特异性抗体(EGFRvⅢ-CD3 bi-scFv)MRSD的方法,同时验证了通过此方法计算得出的FIH剂量在人外周血CD3的理论受体占有率(Theoretical receptor occupancy),来确定这一FIH剂量的安全性。
在本文中,MABEL的计算应用了体外、体内药代动力学/药效学(PK/PD)的数据,包括:在人类与相关物种体内靶细胞上的靶标结合及受体占有率;人类与相关物种靶细胞上的浓度-反应(concentration-response)曲线;相关物种体内的剂量/暴露-反应(dose/exposure-response)曲线等。
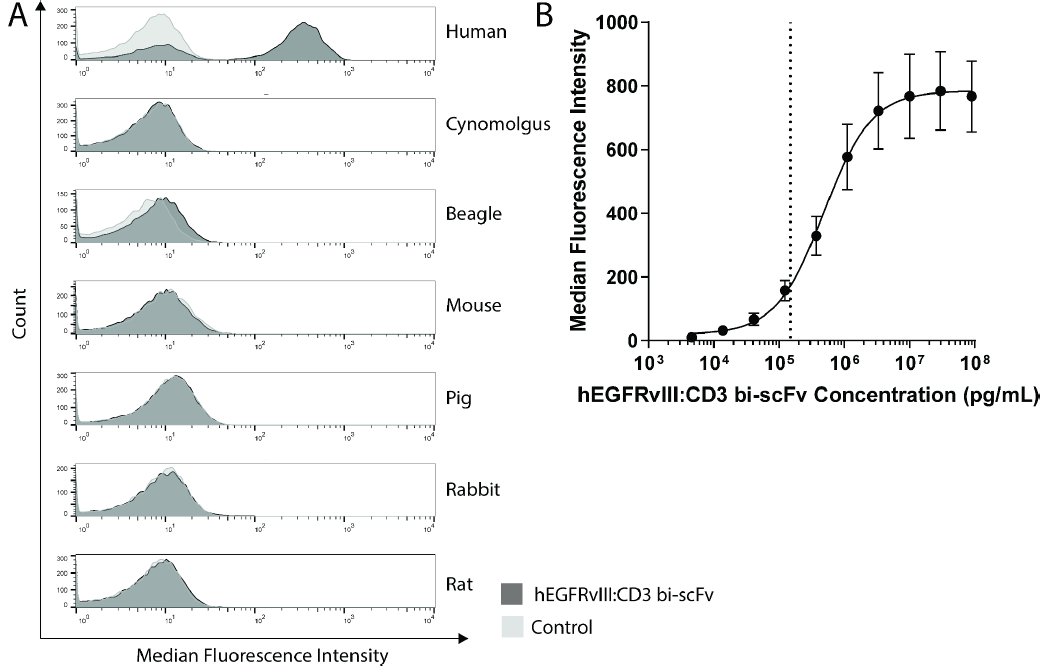
EGFRvⅢ-CD3 bi-scFv与血细胞的结合
研究者使用了流式细胞法,分别检测了EGFRvⅢ-CD3 bi-scFv双特异性抗体与人、比格犬、CD-1小鼠、食蟹猴、Sprague Dawley大鼠、新西兰白兔和猪的外周血单个核细胞(PBMC)的结合能力,来评估EGFRvⅢ-CD3 bi-scFv在细胞水平上与CD3ε的亲和力。实验结果显示(图1),EGFRvⅢ-CD3 bi-scFv只与人的PBMC结合,与其他种类动物的PBMC不结合。与人PBMC结合的EC20为150ng/mL。
图1 流式细胞法检测,在人及相关物种(食蟹猴、比格犬、小鼠、新西兰白兔、大鼠)的PBMC中,EGFRvⅢ-CD3 bi-scFv 只与人的PBMC结合,且EC20为150ng/mL
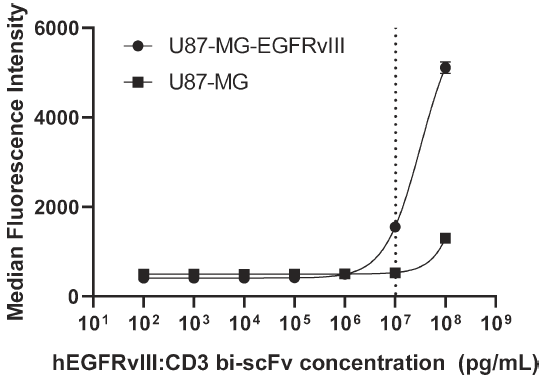
EGFRvⅢ-CD3 bi-scFv与肿瘤细胞的结合
使用流式细胞法,分别检测了EGFRvⅢ-CD3 bi-scFv与野生型人胶质瘤细胞系U87-MG(不表达EGFRvⅢ)及U87-MG-EGFRvⅢ稳转肿瘤细胞系(表达EGFRvⅢ)的结合,用以评估EGFRvⅢ-CD3 bi-scFv与EGFRvⅢ在细胞水平上的结合能力。结果如图2,EGFRvⅢ-CD3 bi-scFv与U87-MG-EGFRvⅢ稳转肿瘤细胞系结合的EC20 为10307 ng/mL,与野生型U87-MG细胞系基本无结合。
图2流式细胞法检测,EGFRvⅢ-CD3 bi-scFv与EGFRvⅢ高表达肿瘤细胞系结合的EC20 为10307 ng/mL
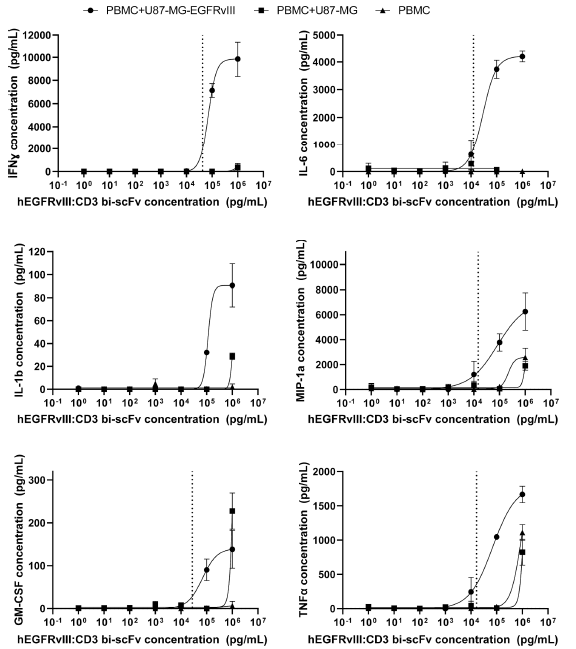
细胞因子释放实验
使用Luminex multiplex platform,分别在PBMC单独存在、PBMC+ U87-MG-EGFRvⅢ细胞(EGFRvⅢ+)、PBMC+ U87-MG细胞(EGFRvⅢ-)条件下,对hEGFRvIII-CD3 bi-scFv介导的人PBMC细胞因子释放进行了检测,使用PMA作为阳性对照。结果显示(图3),hEGFRvIII-CD3 bi-scFv介导人CD3+的细胞产生细胞因子的释放量具有肿瘤抗原依赖性和剂量依赖性,在无EGFRvⅢ抗原存在的情况下,EGFRvIII-CD3 bi-scFv无法诱导细胞因子释放;在有EGFRvⅢ存在的情况下,EGFRvIII-CD3 bi-scFv诱导人PBMC细胞因子IFN-ɣ、IL-6、MIP-1α、GM-CSF和TNFα释放的EC20分别为42.3、12.5、14.7、27.6和16.5 ng/mL。
图3 hEGFRvIII-CD3 bi-scFv介导的人PBMC细胞因子释放呈抗原依赖效应及剂量依赖效应
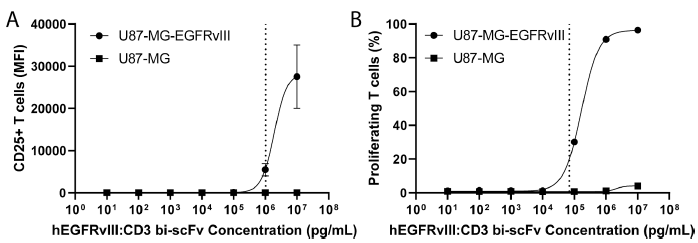
T细胞激活和增殖实验
将EGFRvIII-CD3 bi-scFv在存在肿瘤抗原(U87-MG-EGFRvⅢ)或不存在肿瘤抗原(U87-MG)的条件下与人PBMC共同孵育5天,分别通过检测CD25+T细胞的表达量和CFSE标记细胞的比例来评估EGFRvIII-CD3 bi-scFv激活T细胞及诱导T细胞增殖的能力。结果显示,EGFRvIII-CD3 bi-scFv介导的T细胞激活及T细胞增殖具有抗原特异性和剂量依赖效应。在EGFRvⅢ存在的体系中,EGFRvIII-CD3 bi-scFv激活T细胞和介导T细胞增殖的EC20分别为1021ng/mL、70.2 ng/mL。
图4 EGFRvIII-CD3 bi-scFv在EGFRvIII+体系中介导T细胞激活的EC20为1021ng/mL为,介导T细胞增殖的EC20为70.2 ng/mL
肿瘤细胞杀伤实验
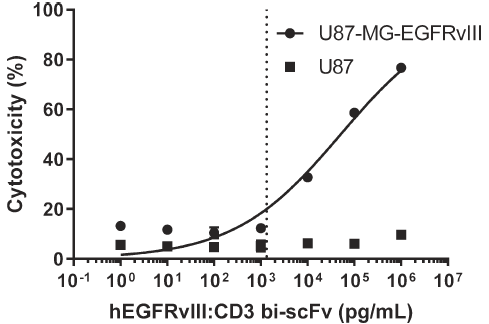
使用铬释放测定法,分别检测了EGFRvIII-CD3 bi-scFv对U87-MG-EGFRvⅢ细胞和U87细胞的杀伤效果,效靶比为20:1。结果显示,EGFRvIII-CD3 bi-scFv对U87-MG-EGFRvⅢ细胞的杀伤效果具有剂量依赖效应,EC20为1.34 ng/mL;对U87野生型细胞几乎无杀伤作用。
图5 EGFRvIII-CD3 bi-scFv对肿瘤细胞的杀伤效果具有肿瘤抗原特异性
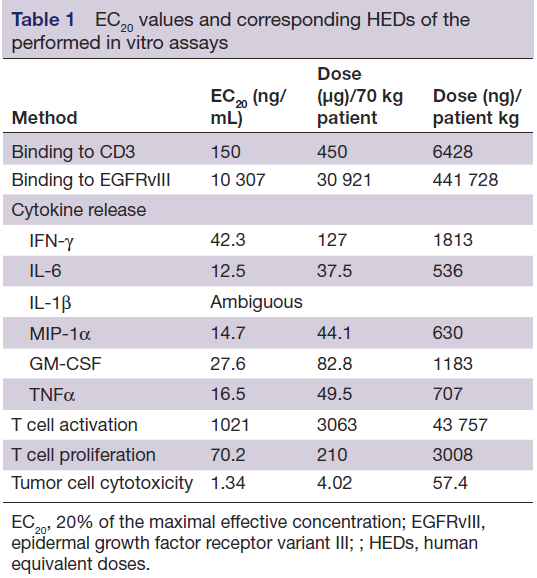
以上全部体外实验结果汇总如表1所示,按照每种体外实验的20%药理活性浓度(20% of the maximal effective concentration,EC20),基于人平均血浆容积为3L、患者平均体重为70kg计算人体初始给药剂量(MRSD)。本系列实验中,最低的EC20来自于肿瘤细胞毒性实验(1.34 ng/mL),相当于每位患者4020 ng或 57.4 ng/kg的起始剂量。
表1 体外实验测定的EC20值及对应的HED值
小鼠体内重复给药、单次给药毒性实验
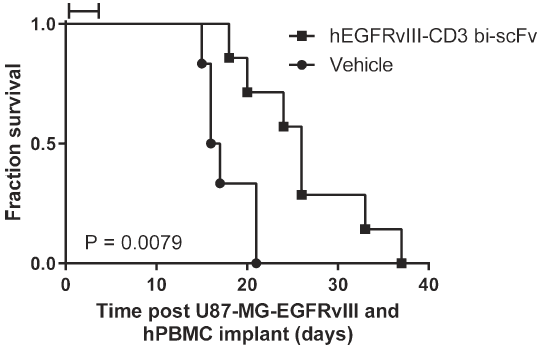
研究人员分别在NSG-人PBMC免疫重建小鼠模型和CD3人源化小鼠模型上进行了体内毒性实验。在移植了U87-MG-EGFRvⅢ胶质瘤细胞和人PBMC的NSG免疫缺陷小鼠中,EGFRvIII-CD3 bi-scFv的给药剂量为2.5 mg/kg,经尾静脉连续给药5天,每日一次。结果显示,给药组的小鼠生存率较溶剂对照组有显著性延长(图6,P=0.0079),证明EGFRvIII-CD3 bi-scFv在此模型上不具有毒性。
在CD3人源化小鼠模型中,研究人员也测试了EGFRvIII-CD3 bi-scFv单次给药10 mg/kg的剂量,同样未观察到显著的毒性反应(数据未给出)。此数据结合该实验室往次实验结果表明,在啮齿动物模型上,每日2.5 mg/kg的给药剂量为安全有效剂量。
图6 EGFRvIII-CD3 bi-scFv在U87-MG-EGFRvⅢ/PBMC-NSG鼠移植模型上可显著提高小鼠生存率
体内有效、安全剂量计算
基于啮齿类模型中每日2.5 mg/kg的安全有效给药剂量,计算人体等效剂量。公式为
CLhu:预期人体清除率;Ceff,ss:预测稳态下有效浓度;Tau:给药间隔
hEGFRvIII-CD3 bi-scFv的结构与博纳吐单抗(Blinatumomab,Blincyto,CD3-CD19双特异性抗体,2014年美国上市)类似,博纳吐单抗在成年人体内的清楚速率为43 mL/h/kg,因此研究者选用这个数据作为CLhu的参考。Ceff,ss为在hCD3转基因小鼠中以5 mg/kg剂量测得的PK数据(0.583 μg/mL)。给药间隔Tau为24小时。
根据此公式,计算出的人体等效剂量为0.6mg/kg,而由体外实验数据计算得出的MABEL剂量为57.4ng/kg,较啮齿类动物模型上药效数据计算出的剂量低10400倍。证实了此剂量(57.4ng/kg)在动物模型上的安全性。
受体占有率计算
靶抗原的理论占有率(receptor occupancy,RO)是药物临床起始剂量选择的重要因素之一。TGN1412 FIH的选择,0.1mg/kg的剂量换算得出的人体外周血中的RO约为90%,对于激动剂而言可能过高。在本文中,研究人员根据两个不同的公式评估了基于MABEL方法计算得出的hEGFRvIII-CD3 bi-scFv给药剂量(57.4ng/kg)在人体中的理论受体占有率,如下所示:
公式1:
公式1基于米氏方程(Michaelis-Menten equilibrium),其中RO为受体占有率,Ab为抗体起始浓度(引用细胞毒性实验EC20,1.34 ng/mL或26.3pM),KD为药物对应抗原的平衡解离常数。
公式2:
公式2为公式1的升级版,由于在已知体系中存在大量靶受体的情况下,由米氏方程计算出的RO可能不准确,因此选用公式2进行RO的计算。其中RO为受体占有率,KD为药物对应抗原的平衡解离常数,TD为总药物浓度(对应细胞毒性实验EC20,1.34 ng/mL或26.3pM),TT为总目标浓度。
TT的计算方法为:
其中n1为每个细胞上CD3受体的数目(6.11×104),n2为每体积中T细胞的数目(1.3×109 L−1),NA为阿伏伽德罗常数。
由公式2计算得出的MABEL剂量的理论受体占有率(引用与CD3ε结合的KD和细胞毒性实验的EC20)为0.17%。对于激动剂而言,根据已知药理学和临床前实验的经验,可接受的RO数据上限约为10%,本研究中基于MABEL剂量计算得出的RO的数据远低于这一限度。但研究人员也说明了这种方法的限制性,由于hEGFRvIII-CD3 bi-scFv为双特异性抗体,该方法无法计算药物同时与表达于不同细胞上的靶抗原(EGFRvIII于肿瘤细胞,CD3于T细胞)结合的理论受体占有率。
本文详细地介绍了一种激动剂型双特异性抗体基于MABEL方法计算FIH的过程及实验思路,通过对比MABEL剂量与临床前体内等效剂量,确定了这种方法的安全性,对于激动型抗体药物的FIH计算具有一定的参考价值。
原创内容未经授权,禁止转载至其他平台。
参考文献
Schaller, Teilo H., et al. "First in human dose calculation of a single-chain bispecific antibody targeting glioma using the MABEL approach." Journal for Immunotherapy of Cancer 8.1 (2020).