来源: 药时空
概述
近年来,由于国家政策的支持,我国的生物医药产业正在成为发展最活跃的产业,医药研发进展可谓日新月异。鉴于药监局的监管政策在不断更新,从事医药研发的企业和人员越来越多。考虑到从事医药研发的新人的增加,有必要将新药研发的基本流程做一个简单梳理。
现代药物的概念除了我们传统意义上的小分子化合物(如阿司匹林,青蒿素),还包括多肽、蛋白质和抗体,(寡)核苷酸,小分子—抗体复合物,还有疫苗。下面以传统的小分子化合物药为例,就新药(主要以1.1类创新药为例)研发从无到有,到最后上市的基本流程做一个概述。
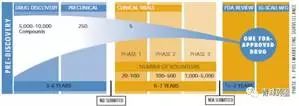
新药的发现(Drug Discovery)
1. 药物作用靶点(target)以及生物标记(biomarker)的选择与确认
早期人们对药物作用靶标认识有限,往往只知道有效,但不知如何起效。比如,百年来,人们知道阿司匹林(aspirin)具有解热、消炎、止痛、抗血栓,甚至抗癌作用。直到1971年,英国人John R. Vane 在《Nature》期刊发文才阐明了Aspirin作用机理为抑制前列腺素合成,并于1982荣获Nobel生理和医学奖。现代生物医学的研究进展,以及人类基因图谱的建立,让人类对疾病的机理了解更加准确,为新药开发提供了明确的方向、具体的靶标。
2. 先导化合物(lead compound)的确定
一旦选定了药物作用的靶标,药物化学家(medicinal chemist)首先要找到一个对该靶标有作用的化合物。这个化合物可以来自天然产物(动物、植物、海洋生物);也可以是根据靶标的空间结构,计算机模拟设计、合成的化合物;还可以根据文献报道或以前其它项目的研究发现。比如,某一类化合物具有作用于该靶标的药理活性或副反应等等。治疗勃起障碍的药物Viagra就是由其副作用开发而成。目前我们常用的方法是跟踪国外研发机构对某一靶标的药物开发,以他们的化合物作为先导,希望设计出更优秀的化合物。
3. 构效关系的研究与活性化合物的筛选
围绕先导化合物,设计并合成大量新化合物,通过对所合成化合物活性数据与化合物结构的构效关系分析,进一步有效的指导后续的化合物结构优化和修饰,以期得到活性更好的化合物。
4. 候选药物(candidate)的选定
通过构效关系研究,几轮优化所有筛选出来的满足基本生物活性的最优化合物,一般就选作为候选药物,进入开发。此时,从事新药发现的药物化学家工作暂告结束。

临床前研究(Pre-clinic toxicology studies)
候选药物确定后,新药研发就进入开发阶段(development),药物开发第一阶段的目标就是完成临床前的毒理学研究,向药监部门提交“实验用新药”(investigational new drug, IND )申请。新药开发需要多学科的协作,比如合成工艺(process chemistry),毒理学,药理学,药代动力学,制剂,等等各学科的协作,另外,所有专业都需要分析化学的支持。
1. 化学、制造和控制(Chemical Manufacture and Control, CMC)
新药开发工作的第一步是原料药合成工艺研发(Process R & D),这是一个不断改进、完善的过程。第一批提供的原料药主要用于毒理研究(100—1000g),要求是越快越好,成本不是主要考虑因素。因此,只要药化路线能够实现毒理批合成,工艺研发部门就会采用。但随着项目的推进,工艺部门会根据需要设计全新合成路线,开发合理生产工艺来满足从I—III期临床用药与商业化的需求;同理,制剂部门首先也会以最简单的形式给药,完成毒理研究,然后不断完成处方工艺研究,开发出商业化的制剂工艺。
2. 药代动力学(Pharmacokinetics, PK)
了解药物在动物体内的吸收、分布、代谢、排泄(ADME),这些数据可以指导临床研究以何种形式给药(口服、吸入、针剂),给药频率与剂量。
3. 安全性药理(Safety Pharmacology)
证明该化合物针对特定目标疾病具有生物活性,同时评估药物对疗效以外的作用,比如可能的副作用,尤其是对心血管、呼吸、中枢神经系统的影响。
4. 毒理研究(Toxicology)
毒理研究种类较多,包括急性毒性、亚急性毒性、慢性毒性、生殖毒性、致癌性、致突变性等。为了加速新药能及早验证是否有疗效,尤其是对一些抗癌药,有些耗时费钱的毒理实验(如致癌性、生殖毒性)是可容許在临床试验阶段再进行。
5. 制剂开发
制剂开发是药物研发的一个重要环节。早期制剂研究并不需要完整的处方开发,所有研究围绕毒理学研究和一期临床时方便给药即可,目的是将候选药物尽快推向临床。随着项目推进,给药方式和处方研究就越来越全面。比如,有的药胃肠吸收很差,就需要开发为注射剂。有的药对在胃酸里面会失去活性,就需要开发为肠溶制剂。有的化合物溶解性不好,也可以通过制剂来部分解决这个问题。
前面这些内容都统称为临床前研究,是药物开发的第一阶段。临床前各个实验的步骤可不是严格按照上述这个顺序展开,而是一个相互包容、相互协调的关系。比如,原料药工艺研发部门,完成毒理批样品合成后,就必须立即开展合成路线的选择,开发新的合成工艺,提供足够量的原料药以满足制剂部门制剂研究用原料药和9-12个月后开展I期临床用药的需求。
因此,项目推进是否顺利,就看各专业间的协调与配合是否密切了。
临床研究(Clinical studies)
当一个化合物通过了临床前试验后,需要向药监部门(C)FDA提交新药临床研究申请(IND),以便可以将该化合物应用于人体试验。新药临床研究申请需要提供先前试验的材料;以及计划将在什么地方,由谁以及如何进行临床试验的说明;新化合物的结构;给药方式;动物试验中发现的所有毒性情况;该化合物的制造生产情况。所有临床方案必须经过伦理审评委员会(Institutional Review Board,IRB)的审查和通过,每年还必须向FDA和IRB汇报一次临床试验的进程和结果。在美国,如果在提交申请后30天内FDA没有驳回申请,那么该新药临床研究申请即被视为有效,可以进行人体试验。在中国则需要获得CFDA正式批准,方可进入临床。
Ⅰ期临床试验
在新药开发过程中,将新药第一次用于人体以研究新药的性质的试验,称之为Ⅰ期临床试验。这一阶段的临床试验一般需要征集20-100名正常和健康的志愿者(对肿瘤药物而言通常为肿瘤病人,但人数更少),在严格控制的条件下,给不同剂量(随着对新药的安全性了解的增加,给药的剂量也逐渐提高,并可以多剂量给药)的药物试验于健康志愿者,住院以进行24小时的密切监护,仔细监测药物的血液浓度、排泄性质和任何有益反应或不良作用,以评价药物在人体内的性质。同时也要通过这一阶段的临床试验获得其吸收、分布、代谢和排泄以及药效持续时间的数据和资料;以及药物最高和最低剂量的信息,以便确定将来在病人身上使用的合适剂量。可见,Ⅰ期临床试验是初步的临床药理学及人体安全性评价试验,目的在于观测人体对新药的耐受程度和药代动力学,为制定给药方案和安全剂量提供依据。
Ⅱ期临床试验
为了证实药品的治疗作用的,就必须在真正的病人身上进行临床研究,即Ⅱ期临床试验。Ⅱ期的临床试验通常需要征集100-500名相关病人进行试验。其主要目的是获得药物治疗有效性资料。
将试验新药给一定数量的病人志愿者,评价药物的药代动力学和排泄情况。这是因为药物在患病状态的人体内的作用方式与健康志愿者是不同的,对那些影响肠、胃、肝、和肾的药物尤其如此。Ⅱ期临床试验一般通过随机盲法对照试验(根据具体目的也可以采取其他设计形式),对新药的有效性和安全性作出初步评价,并为设计Ⅲ期临床试验和确定给药剂量方案提供依据。
Ⅲ期临床试验
当一个新药推进到三期临床,原料药和制剂工艺研究也推进到了相应的阶段。三期临床用药以商业化生产工艺提供临床用药。一般来讲,商业化生产的原料药生产工艺应该考虑以下因素:产品质量,生产安全性,生产成本,环境影响,生产的稳定性和可持续性。
Ⅲ期的临床试验通常需 1000-5000名临床和住院病人,在医生的严格监控下,进一步获得该药物的有效性资料和鉴定副作用,以及与其他药物的相互作用关系。该阶段试验一般将对试验药物和安慰剂(不含活性物质)或已上市药品的有关参数进行对照和双盲法试验(医生和病人都不知道自己吃的是新药、老药或安慰剂),在更大范围的病人志愿者身上,进行扩大的多中心临床试验。最后,根据严格统计学数据分析,进一步评价药物的有效性和耐受性(或安全性),决定新药是否优于(superior)或不差于(not inferior)市场现有的“老药”。Ⅲ期临床试验是治疗作用的确证阶段,也是为药品注册申请获得批准提供依据的关键阶段,该阶段是临床研究项目的最繁忙和任务最集中的部分,无疑是整个临床试验中最重要的一步。三期临床研究往往持续好几年。
除了对成年病人研究外,还要特别研究药物对老年病人,有时还要包括儿童的安全性。一般来讲,老年病人和危重病人所要求的剂量要低一些,因为他们的身体不能有效地清除药物,使得他们对不良反应的耐受性更差,所以应当进行特别的研究来确定剂量。而儿童人群具有突变敏感性、迟发毒性和不同的药物代谢动力学性质等特点,因此在决定药物应用于儿童人群时,权衡疗效和药物不良反应应当是一个需要特别关注的问题。在国外,儿童参加的临床试验一般放在成人试验的Ⅲ期临床后才开始。如果一种疾病主要发生在儿童,并且很严重又没有其他治疗方法,美国食品与药品管理局允许Ⅰ期临床试验真接从儿童开始,即在不存在成人数据参照的情况下,允许从儿童开始药理评价。我国对此尚无明确规定。
上述任何一步反馈得到的结果不好,都有可能让一个候选药物胎死腹中。最悲惨的结果可能是这个项目就直接被取消了。2007年,Merck有四个三期临床药物失败。
能够通过全部3期临床评价而上市的新药越来越少,部分原因是开发出比市场上现有药物综合评价更好的新药越来越难。而一个药物从源头研发到3期临床是一个耗资巨大的过程。公开数据表明,平均下来一个新药要花费约为十亿美金(1 billion US dollars)。正是因为药物研发的耗资巨大,大公司花不起那么多钱同时展开多个项目研究,小公司又没有那么多的财力完成药物研发的全部流程。现在的药物研发的一个趋势是,小公司反而能够更好的找准市场上的空缺,开发出在临床前研究阶段具有良好表现的候选药物。这时大公司通过并购小公司或者购买专利(或者使用权),将这个项目买过来继续开发。如果是购买专利的情况,则会根据这个项目最后能够进展到哪个阶段,完成后相应得再支付给小公司一笔“奖金”,叫做milestone。
新药申请(New drug application, NDA)
完成所有三个阶段的临床试验并分析所有资料及数据,药物的安全性和有效性得到了证明,新药持有人则可以向药监部门(C)FDA提交新药申请。新药申请需要提供所有收集到的科学资料。通常一份新药申请材料可多达100000 页,甚至更多!按照法规,FDA应在6个月内审评完新药申请。但是由于大部分申请材料过多,而且有许多不规范,因此往往不能在这么短的时间内完成。中国药监局也在努力改进工作,期望缩短审批时间。
批准上市
新药申请一旦获得药监部门批准,该新药即可正式上市销售,供医生和病人选择。但是新药持有人还必须定期向药监部门呈交有关资料,包括该药物的副作用情况和质量管理记录。对于有些药物药监部门还会要求做第四期临床试验,以观测其长期副作用情况。
如果能够走到这一步,那么暂时可以说是大功告成了。从最开始的备选化合物走到这一步的药物寥寥无几。但是批准上市了并不代表这个药物就高枕无忧了。因为还有后面一步。

IV期临床研究(药物上市后监测)
药物在大范围人群应用后,需要对其疗效和不良反应继续进行监测。药监部门要求根据这一阶段的监测结果来修订药物使用说明书。这一阶段研究还会涉及到的一些内容有,药物配伍使用的研究,药物使用禁忌。如果批准上市的药物在这一阶段被发现之前研究中没有发现的严重不良反应,比如显著增加服药人群心血管疾病发生率之类的,药物还会被监管部门强制要求加注警告说明,甚至下架。如Merck的抗关节炎药物Vioxx 因增加心血管疾病风险于2004 年“主动”撤离市场。
总之,新药研发是一个高风险,高投入,当然也是高回报的行业。研发周期长,涉及多学科、多专业的密切配合与协调。如果一个新药研发机构各专业职能部门配套合理,各专业人员能够做好各自的本职工作,又注重各专业间的配合和与药监局的及时沟通,新药研发管理并不复杂,只要有一个负责任的项目管理员,根据约定的项目关键时间节点及时协调各个职能部门的工作,项目就可按计划有序进行。
返回最上面
|




